Drop-seq: single-cell sequencing in large scale
Drop-seq greatly improves the through-put of single-cell sequencing where transcriptome data of thousands of cells could be generated. This post highlights 3 features, 1 tech issue, 1 aspect of downstream analysis.
STAMP
STAMP: single-cell transcriptome attached to microparticle. Due to the primer bead design, i.e. Cell barcode + UMI, the source of single-cell’s transcriptome would not be ambiguous and rather clearly determined.
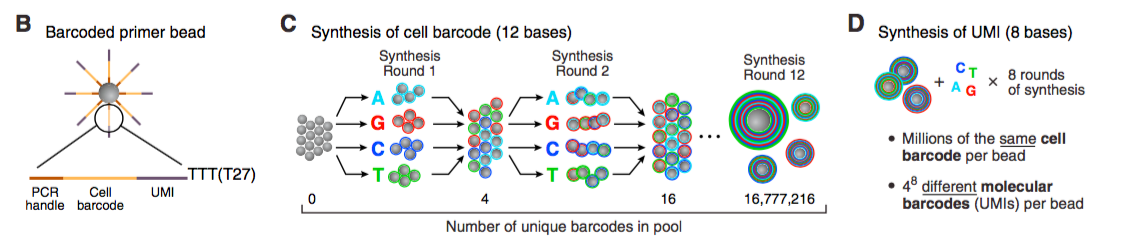
Not compatible for pre-transcriptional layer, but …
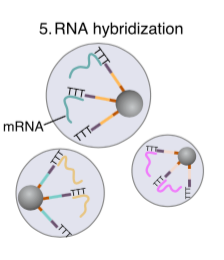
Hybridization
The poly-T end of the primer bead is responsible to attract the poly-A tail of mRNA. It is like Oligo dt primer in reverse transcription. Therefore only the matured mRNA will be captured. Any study at pre-transcriptional layer cannot use Drop-seq tech.
However, oligo dt primer is not the only choice during cDNA library construction, but we still have random primer. In the same way, probably the poly-T end could be abandoned, or replaced by further rounds synthesis of UMI, because generating UMI is nothing but adding random A/G/C/T to the end of cell barcode.
Yes, absolute amount of transcriptome
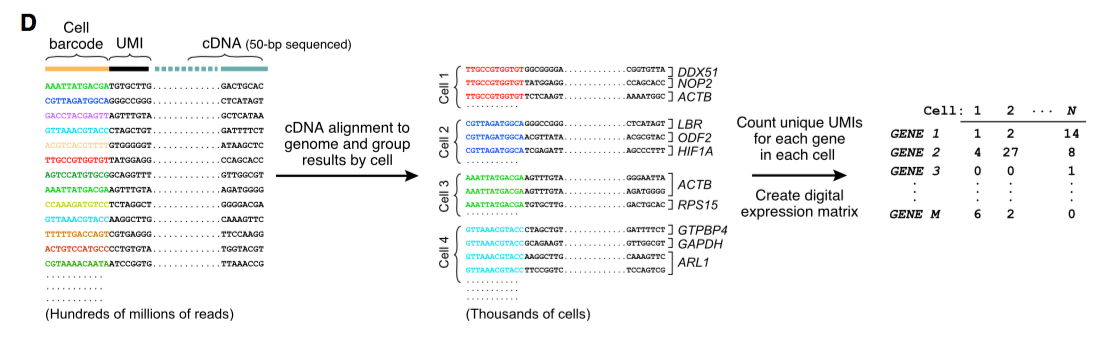
Counting the unique UMIs for each gene in each cell generating the absolute amount of transcripts expression. Yes, no more RPKM.
Due to the PCR amplification with STAMPs as templates, generated sequencing signals are robust and in the same time UMIs are expected to be duplicated. Only unique ones are taken into account for yielding the digital expression matrix. Cooking the reads into data matrix is done by a Java software developed by Jim Nemesh (see analysis manual here: http://mccarrolllab.com/dropseq/)
Single particle binds ≥ 1 cells? It happens.
Referring to Fig-3, it happens when the concentration of cells is high thus one particle could possibly attach more than one cells.
Classifying cells using tSNE
The paper first performed PCA on original full features to abstract 32 significant PCs, then run tSNE on these PCs which further reduce dimension to 2D thus friendly for scatter plot.
Classification is implemented by Seurat, a R language package developed and maintained by Rahul Satija lab. It follows GPL 3.0 license and I have implemented patches, honflueR (see github: https://github.com/Puriney/honfleuR). Without violating seurat analysis context, it speeds up original functions and incorporates several new imputation strategies.